










La Esterilización está destinado a generar Dispositivos médicos reutilizables (DMR) libres de microorganismos viables
![]() Los Priones no son microorganismos y son más resistentes que los microorganismos convencionales.
Los Priones no son microorganismos y son más resistentes que los microorganismos convencionales.
La esterilización de DMR se basa en 3 conceptos claves
- Nivel de aseguramiento de la esterilidad (SAL): No es posible controlar sistemáticamente la ausencia total de microorganismos viables. El objetivo de la esterilización es limitar la probabilidad de sobrevivencia de los microorganismos a un nivel muy bajo. Para los DMR, la probabilidad o nivel de aseguramiento de la esterilidad (SAL) se expresa como: no mayor que 1 microorganismo viable en una cantidad de un millón de artículos esterilizados (o SAL 10-6 ).
- Sobreletalidad: Se desconoce la cantidad, naturaleza y ubicación de los microorganismos que pueden estar presentes en un DMR después de la limpieza. Los procesos de esterilización de los DMR deben demostrar su capacidad para inactivar un inóculo altamente concentrado, mayor a 1 millón de microorganismos de prueba. Los microorganismos de prueba se seleccionan con una alta resistencia al proceso de esterilización. Este margen conservador, por encima del nivel más alto de contaminación de la vida real, se denomina método de sobreletalidad.
- Compatibilidad:los DMR siguen siendo completamente funcional y seguros para su uso después de la esterilización. La compatibilidad es verificada por el fabricante del DMR, que determina el número máximo de ciclos de esterilización a los que se puede exponer un DMR, antes de ser desechado o reparado.
Varios métodos han sido propuestos por normas internacionales para demostrar la capacidad de un proceso de esterilización dado, para alcanzar el SAL mediante el método de sobreletalidad. Uno de ellos es el método de Sobreletalidad de medio ciclo, que se describe a continuación:
![]() La esterilización por radiación (ionizante – gamma, rayos electrónicos o rayos X de alta energía, o ultravioleta no ionizante (UV)) no se usan comúnmente para el reprocesamiento de DMR en centros de salud y no se tratarán en las presentes directrices.
La esterilización por radiación (ionizante – gamma, rayos electrónicos o rayos X de alta energía, o ultravioleta no ionizante (UV)) no se usan comúnmente para el reprocesamiento de DMR en centros de salud y no se tratarán en las presentes directrices.
Los métodos de esterilización no terminal cumplen con los criterios SAL. Sin embargo, a diferencia de la esterilización terminal, los DMR no se protegen por empaques. La esterilización por vapor de uso inmediato (anteriormente denominada esterilización flash) es un ejemplo de proceso de esterilización no terminal.
Todos los procesos de esterilización requieren precauciones de salud y seguridad ocupacional
- La alta presión en las cámaras de esterilización con vapor, implica el control periódico de la integridad de la cámara, de acuerdo con las regulaciones o recomendaciones.
- En la esterilización con vapor a alta temperatura y la esterilización por calor seco se corre el riesgo de sufrir quemaduras. Los DMR tienen tiempo de enfriamiento y los operadores deben usar guantes.
- Todos los productos químicos usados para baja temperatura son tóxicos a diferentes niveles (por eso su eficacia en los microorganismos). Los controles periódicos verifican la ausencia de fugas. Antes de acceder a un DMR, los residuos se eliminan a los niveles definidos por las regulaciones de seguridad y salud ocupacional aplicables.
En el mercado se ofrecen varios tamaños y configuraciones de esterilizadores.
- Los esterilizadores grandes se utilizan en las instalaciones de una central de esterilización. A menudo son sistemas de doble puerta (con entrada y salida).
- Los esterilizadores de sobremesa y de una puerta se utilizan en clínicas ambulatorias, dentales y rurales.
La elección del método de esterilización se realiza de acuerdo con los pricipios de Clasificación de Spaulding y las regulaciones o recomendaciones locales aplicables.
Las preferencias o tendencias comunes se pueden resumir de la siguiente manera:
- La esterilización terminal se prefiere a la esterilización no terminal para los DMR que entran en cavidades estériles.
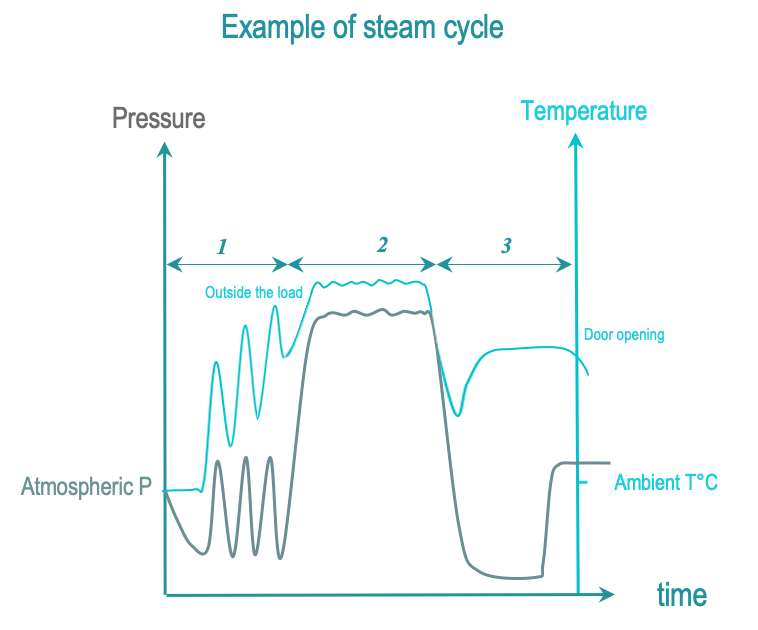
- La esterilización por vapor se recomienda para DMR compatibles con humedad y calor. Los ciclos de vapor más eficientes son los de 132 ° C (270 ° F) o 134 ° C. El tiempo de meseta requerido o recomendado varía desde 3 minutos hasta 18 minutos según la normativa aplicable.
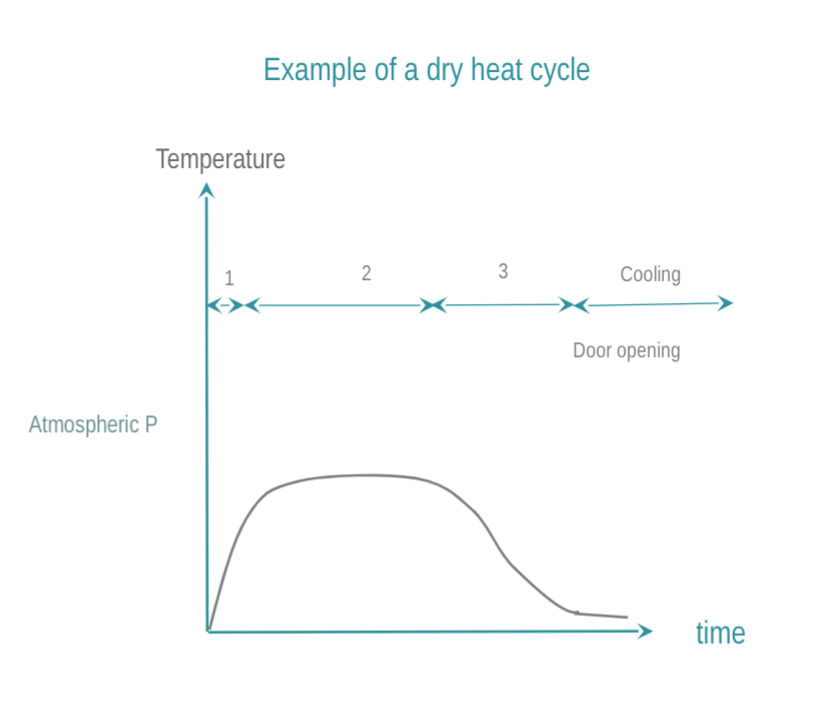
- La esterilización por calor seco está prohibida en un número creciente de países debido a su propiedades fijadoras y malos resultados en comparación con la esterilización por vapor.
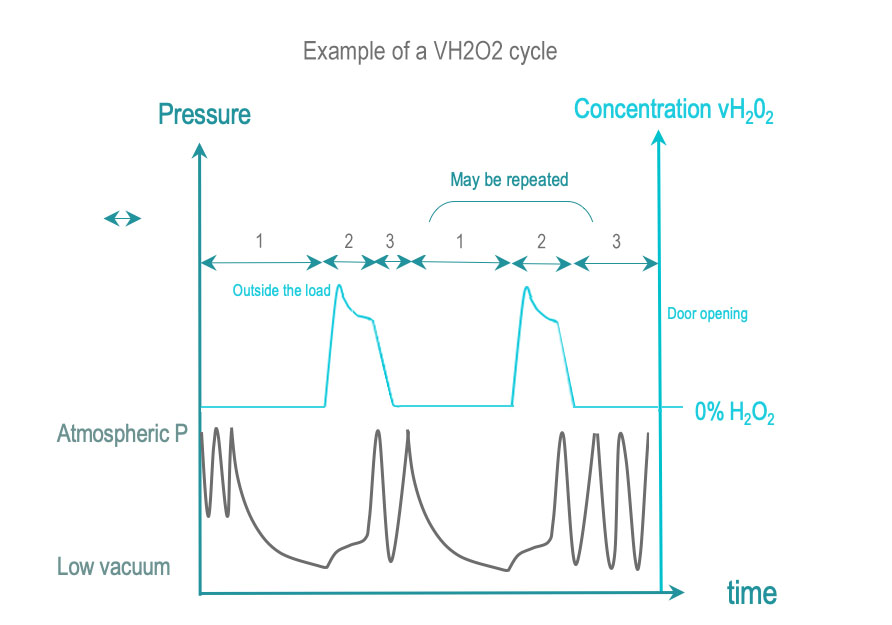
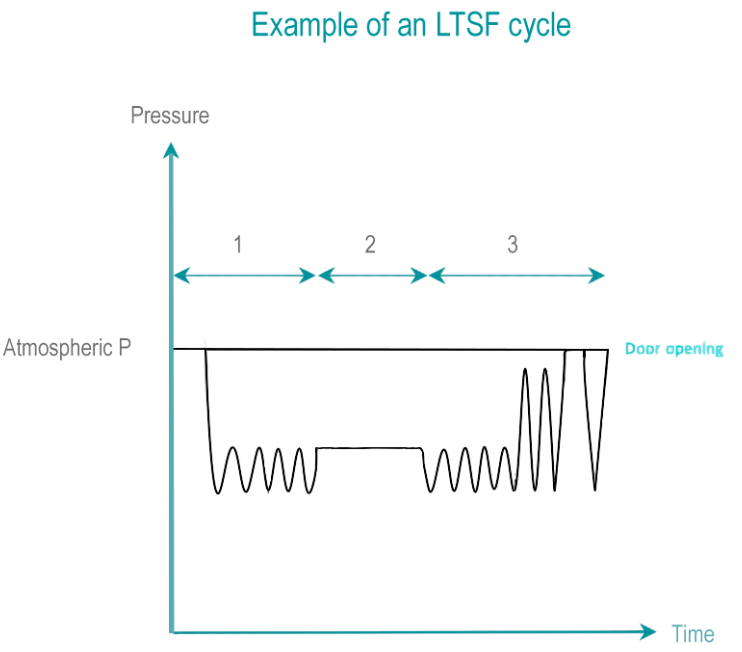
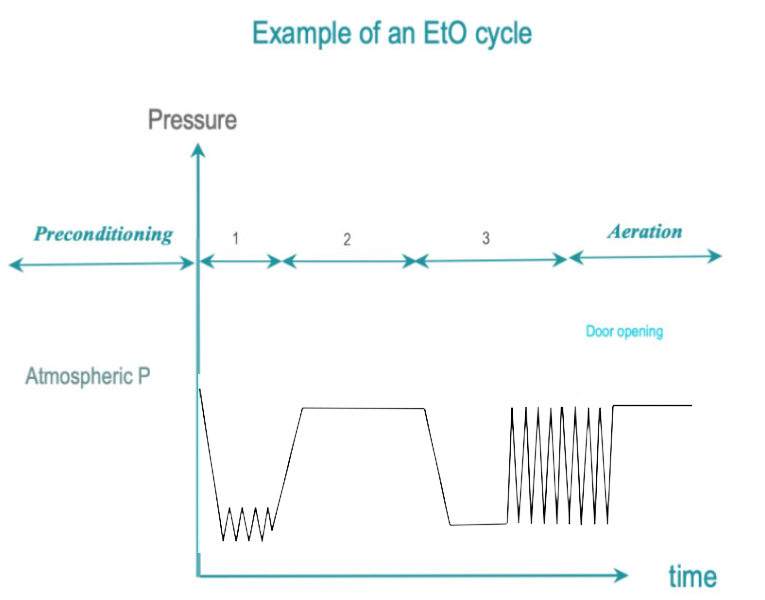
- Para los DMR sensibles al calor, la elección del método de esterilización terminal a baja temperatura (LTS) sigue las instrucciones de uso de los fabricantes del DMR y puede estar influenciado por convenciones, pautas o regulaciones regionales. El peróxido de hidrógeno vaporizado (VH2O2) es el método LTS más aceptado. El formaldehído de vapor a baja temperatura (LTSF) no se utiliza en países con fuertes regulación de priones, debido a sus propiedades fijadoras. El uso de EtO depende del país debido a las propiedades fijadoras, el largo tiempo de aireación y las restricciones de seguridad y salud ocupacional.
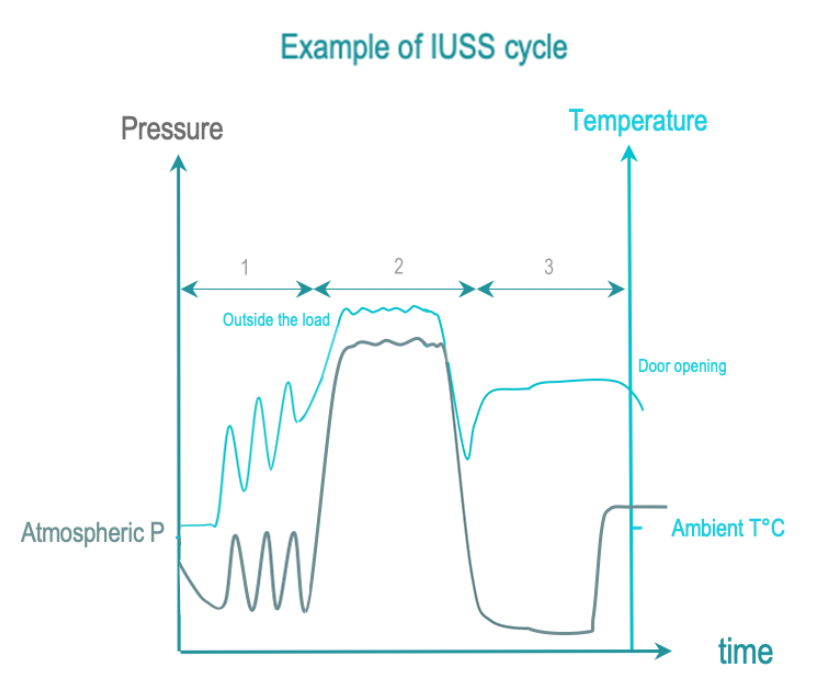
- En algunos países no se utiliza la IUSS no terminal. Sigue siendo tolerado en otros, pero generalmente con recomendaciones para usar esterilización terminal con vapor.
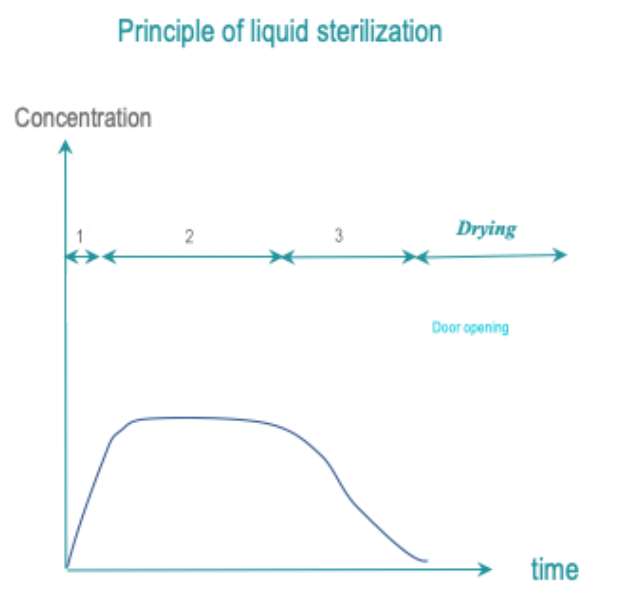
- La aceptación de procesos esterilizantes líquidos no terminales es dependiente de la región. En un proceso con líquido esterilizante, el DMR se sumergen en una solución consiguiendo 10-6 SAL. Luego se enjuagan los DMR. Los DMR no están protegidos por un embalaje. Por tanto, los procesos de esterilización líquida no son terminales.
El nivel de flexibilidad que se deja al usuario para elegir el método de esterilización, depende de las normas o regulaciones locales. Por ejemplo, en algunos países se utiliza vapor, excepto cuando no está permitido por la instrucciones de uso del fabricante del DMR. En otros países, esterilización a baja temperatura se usa o se tolera para dispositivos compatibles con vapor que se sabe que se deterioran por la exposición repetida al vapor (por ejemplo, ópticas).
La conformidad de un proceso de esterilización a normas internacionales puede ser requerido por las regulaciones locales aplicables.
![]() Las Normas específicas están disponibles para vapor1,2,3,4,5 para calor seco6, para EtO7,8 y LTSF9,10. La ISO 14937 se utiliza para VH2O2 (actualmente se están desarrollando normas específicas) 12,13. No existen normas internacionales directamente aplicables para los procesos de esterilización con líquidos y IUSS.
Las Normas específicas están disponibles para vapor1,2,3,4,5 para calor seco6, para EtO7,8 y LTSF9,10. La ISO 14937 se utiliza para VH2O2 (actualmente se están desarrollando normas específicas) 12,13. No existen normas internacionales directamente aplicables para los procesos de esterilización con líquidos y IUSS.
![]() No existen normas internacionales para IUSS.
No existen normas internacionales para IUSS.
![]() Las regulaciones o normas locales pueden requerir o recomendar que la esterilización se lleve a cabo en un departamento de esterilización centralizado, prohibiendo así la esterilización en el lugar de uso. Los quirófanos no suelen estar equipados ni organizados para limpiar y secar DMR de forma como lo hace el departamento de esterilización centralizado.
Las regulaciones o normas locales pueden requerir o recomendar que la esterilización se lleve a cabo en un departamento de esterilización centralizado, prohibiendo así la esterilización en el lugar de uso. Los quirófanos no suelen estar equipados ni organizados para limpiar y secar DMR de forma como lo hace el departamento de esterilización centralizado.

Recomendaciones clave de WFHSS para limpieza y desinfección
IUSS to be replaced by steam sterilization
Go to IUSS sterilization →
1 of 16 Esterilizante líquidoTo be evaluated by WFHSS
Go to Liquid sterilization →
2 of 16 VaporSteam 134°C or 132°C preferred when allowed by RMD IFU
Go to Steam sterilization →
3 of 16 LTSFCycle according to RMD IFU
Go to Low temperature steam formaldehyde →
4 of 16 VH2O2Cycle according to RMD IFU
Go to Vaporized H2O2 →
5 of 16 EtOCycle according to RMD IFU
Go to Ethylene Oxide →
6 of 16 DMR limpio, seco y empaquetadoNon packaged for non terminal sterilization
Go to choice of sterilization process →
7 of 16 Dispositivo médico estérilNon packaged RMD for immediate use when non terminal sterilization
Packaged RMD for storage when terminal sterilization
Go to choice of sterilization process →
Terminal sterilization preferred to
non terminal
Go to choice of sterilization process →
9 of 16 Esterilización no terminalThe RMD is not protected by a packaging and must be immediately used after sterilization
Go to choice of sterilization process →
10 of 16 +Terminal sterilization preferred
Go to recommendation of WFHSS for sterilization →
11 of 16 +Steam sterilization at 132°C or 134°C preferred when allowed by RMD IFU
Go to recommendation of WFHSS for sterilization →
12 of 16 +Visual control and routine controls
Go to Sterilization and quality management →
13 of 16 +According to RMD IFU
Go to choice of sterilization process →
14 of 16 +Steam sterilization at 132°C or 134°C preferred when allowed by RMD IFU
Go to recommendation of WFHSS for sterilization →
15 of 16 +Visual control and routine controls
Go to Sterilization and quality management →
16 of 16

