




![]() 朊毒体不是微生物,比普通的微生物更具抗力。
朊毒体不是微生物,比普通的微生物更具抗力。
可重复使用的医疗器械(RMD)的灭菌基于 3 个关键概念
- 无菌保证水平 (SAL):不可能系统地控制完全没有存活微生物。灭菌的目标是将微生物存活的可能性限制在一个非常低的水平。对于 RMD,该可能性或无菌保证水平 (SAL) 表示为:在 100 万个已灭菌物品中不超过有 1 个存活微生物(或 10-6 SAL)。
- 过度杀灭:可能存在于清洁后的 RMD 上的微生物的数量、性质和位置不能明确。 RMD 灭菌过程必须证明其能够灭活超过 100 万数量级的测试微生物的高浓度接种物。选择测试微生物是基于它们对灭菌过程具有高抗力。这种高于实际污染最高水平的保守裕量被称为过度杀灭法。
- 兼容性:RMD 在灭菌后能保持完整的功能和安全使用。兼容性由 RMD 制造商检查,该制造商决定了 RMD 在被丢弃或维修之前,可以暴露的最多的灭菌周期数。
国际标准提出了多种方式来证明给定灭菌过程应用过度杀灭方法达到 SAL 的能力。 其中之一是半周期过度杀灭方式,描述如下:
使用已知对灭菌过程具有高抗力的微生物(通常是细菌芽孢)进行测试。
灭活动力学是线性的,则杀灭 106 微生物的时间或剂量加倍就会产生 10-6 SAL。例如,D 值为 2 分钟的灭菌过程在 12 分钟内灭活 6 log,则在 24 分钟内可达到 SAL。 一旦对过程进行了表征,就必须验证 是否可以有效灭菌RMD。将 106 接种物放置在 RMD 上或内部最难灭菌的位置。 将包装好的RMD放入具有代表性的具有挑战性的负载中。 RMD 制造商的 IFU 指明废置或维修前的最多使用次数。 用于常规控制时,接种物可以放置在过程挑战装置 (PCD) 中 | 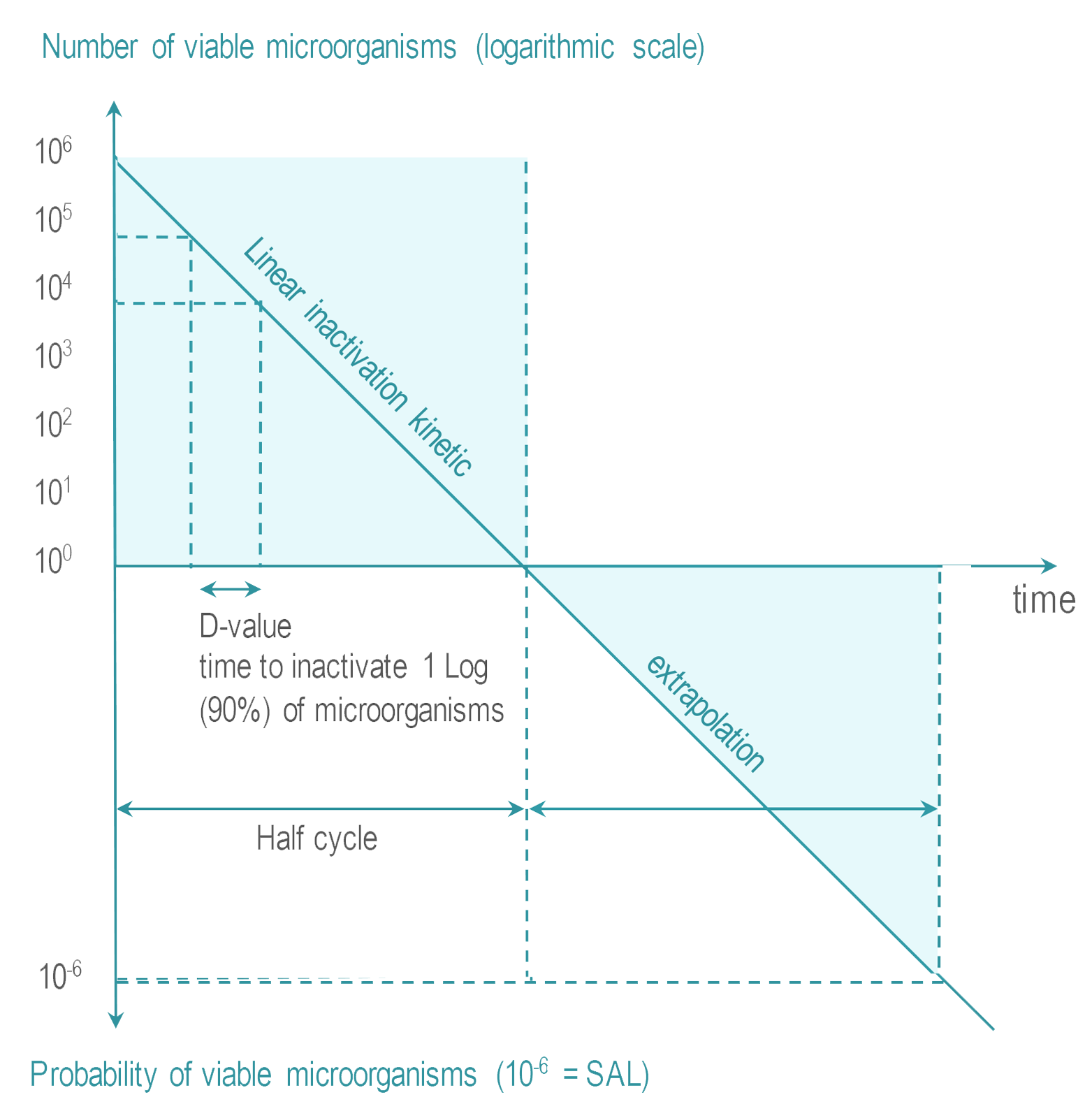 |
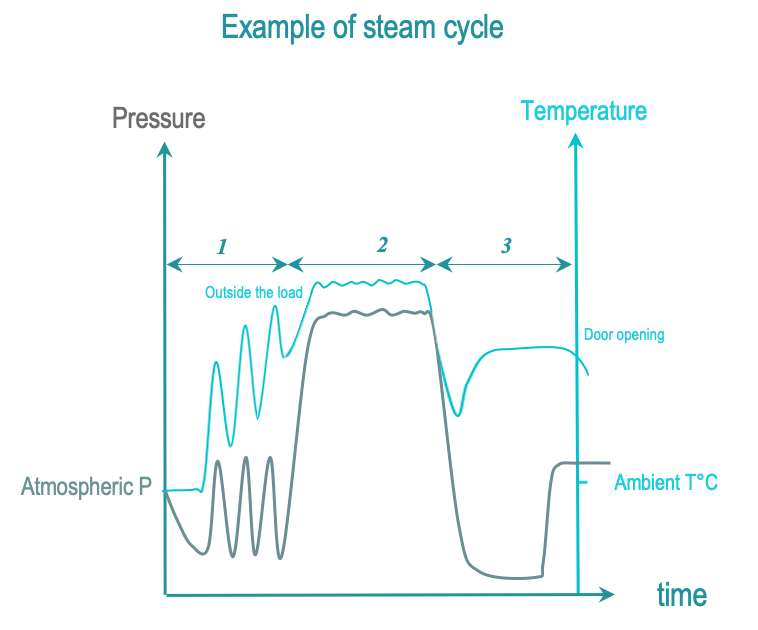
蒸汽灭菌是最常见的灭菌方法。 也称为湿热灭菌或饱和蒸汽灭菌),
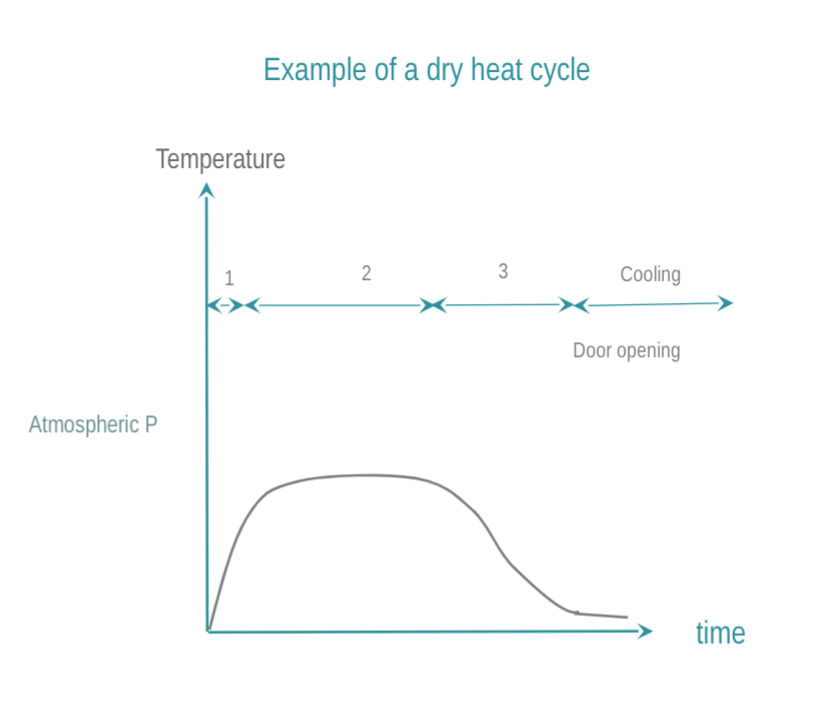
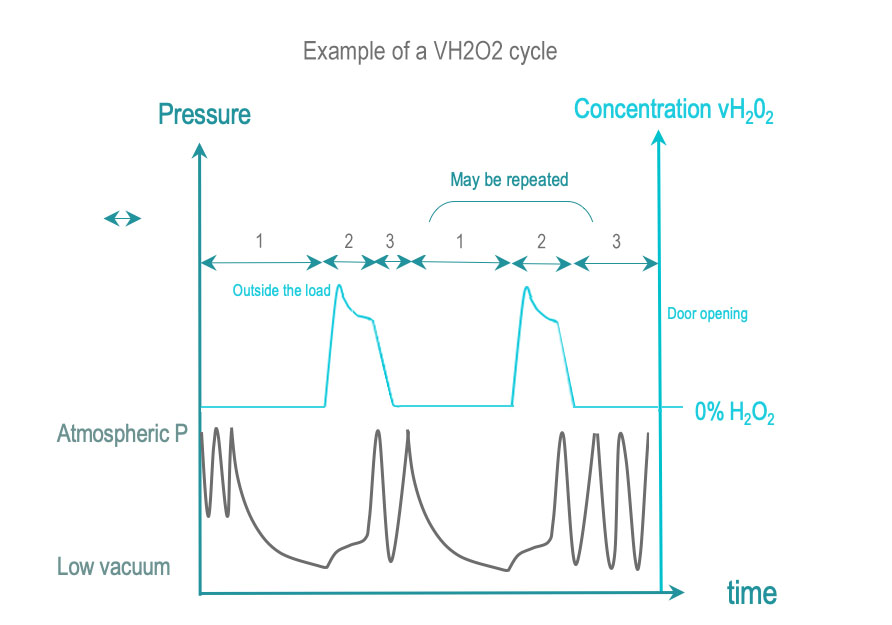
在干热灭菌中,RMD 暴露在干燥的热空气中。 低温灭菌 (LTS) 适用于不耐高温的 RMD。 目前的低温灭菌因子有:环氧乙烷 (EtO)、蒸汽甲醛 (LTSF)、汽化过氧化氢 (VH2O2) 和臭氧 (O3)。 在达到目标 SAL 所需的时间内,RMD 在受控的温度、湿度和/或压力条件下暴露于最低浓度 (Cc) 的灭菌因子。
|
![]() 辐射灭菌(电离 – 伽马、电子束或高能 X 射线,或非电离紫外线 (UV))通常不用于医疗机构中 RMD 的再处理,本指南中将不讨论。
辐射灭菌(电离 – 伽马、电子束或高能 X 射线,或非电离紫外线 (UV))通常不用于医疗机构中 RMD 的再处理,本指南中将不讨论。
非最终灭菌方法满足 SAL指标。然而,与最终灭菌不同,RDM 不受包装保护。即时使用蒸汽灭菌(以前称为快速灭菌)是非最终灭菌过程的一个例子。
实施所有灭菌过程都需要职业健康和安全的预防措施。
- 蒸汽灭菌器腔体中的高压,要求根据适用法规或专业指南对腔体完整性进行定期控制。
- 蒸汽和干热灭菌的高温有灼伤危险。 RMD需要有时间冷却,操作员应戴上手套。
- 所有低温化学品都具有不同程度的毒性(这就是它们对微生物有效的原因)。定期控制检查是否有泄漏。在接触 RMD 之前,残留物应减少至相应职业健康和安全法规规定的水平。
市场上有各种尺寸和配置的灭菌器。
- 中央灭菌部门使用大型灭菌器。 通常是双门(直通)类型。
- 台式、单门灭菌器用于门诊、牙科和农村诊所。
根据 Spaulding 分类原则和适用的当地法规或专业指南选择灭菌方法。
常见的选择原则或技术趋势可概括如下:
- 当RMD 进入人体无菌腔道时,最终灭菌优于非最终灭菌。
- 对于耐受湿热的RMD,推荐使用蒸汽灭菌。最有效的蒸汽周期是 132°C (270°F) 或 134°C 的周期。根据适用法规,要求或推荐的维持时间从 3 分钟到 18 分钟不等。
- 干热灭菌由于其固定蛋白质性质和与蒸汽灭菌相比的性能较差,而在越来越多的国家被禁止。
- 对于热敏 RMD,终末低温灭菌 (LTS) 方法的选择遵循 RMD 制造商的 IFU,可能会受到地区公约、指南或法规的影响。汽化过氧化氢 (VH2O2) 是最广泛接受的 LTS 方法。低温蒸汽甲醛 (LTSF) 因其固定蛋白质特性而未在朊毒体监管严格的国家/地区使用。由于固定特性、解吸时间长以及职业健康和安全限制,EtO 的使用取决于国家/地区。
- 部分国家不使用非终末IUSS。但它在其他国家中仍然允许,但通常推荐使用终末蒸汽灭菌。
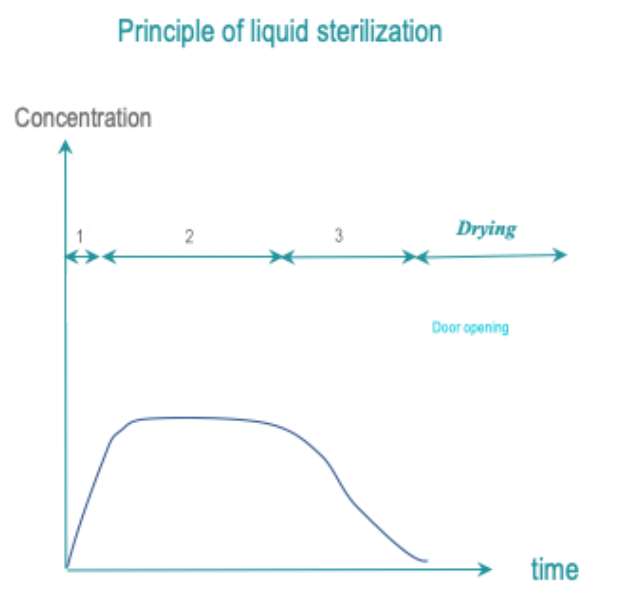
- 非终末液体灭菌过程的接受程度取决于地区。在液体灭菌过程中,RMD 被浸入产生 10-6 SAL 的溶液中。然后接着冲洗 RMD。 RMD 不受包装保护。因此,液体灭菌剂过程是非终末的。
用户可以灵活选择灭菌方法,这取决于当地法规或指南。例如,在某些国家/地区会使用蒸汽,除非 RMD 制造商的 IFU 不允许。在一些国家/地区,LTS 用于耐受蒸汽的器械(例如,光学器件)避免反复暴露于蒸汽而造成性能下降。
当地法规可能要求灭菌过程同时符合国际标准。
![]() 蒸汽1、2、3、4、5 干热6、EtO7、8 和LTSF9、10 有专用标准。 ISO 14937 用于 VH2O2(目前正在制定专用标准)12,13。 没有直接适用于液体灭菌剂过程和 IUSS 的国际标准。
蒸汽1、2、3、4、5 干热6、EtO7、8 和LTSF9、10 有专用标准。 ISO 14937 用于 VH2O2(目前正在制定专用标准)12,13。 没有直接适用于液体灭菌剂过程和 IUSS 的国际标准。

WFHSS 清洁和消毒的关键建议
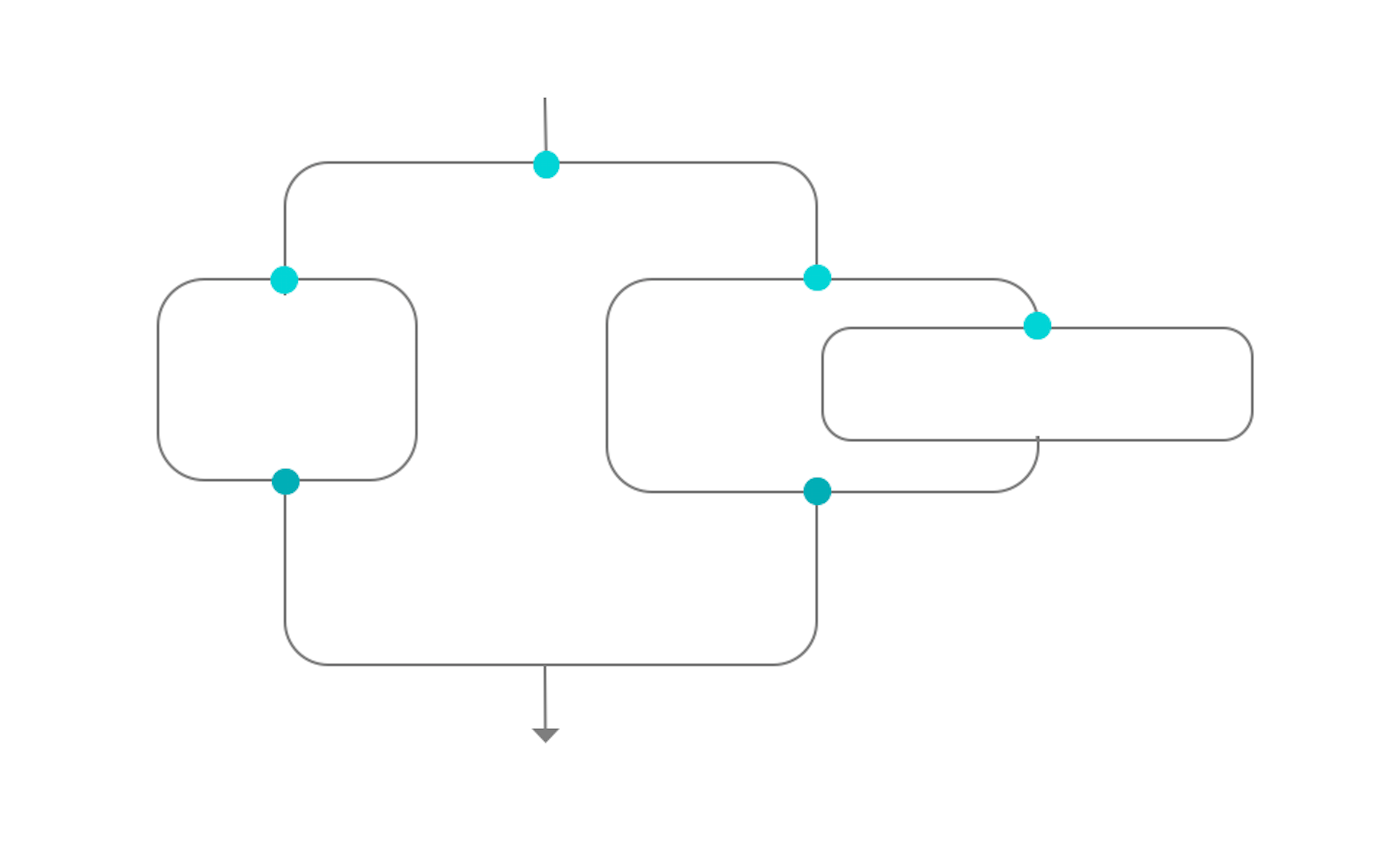
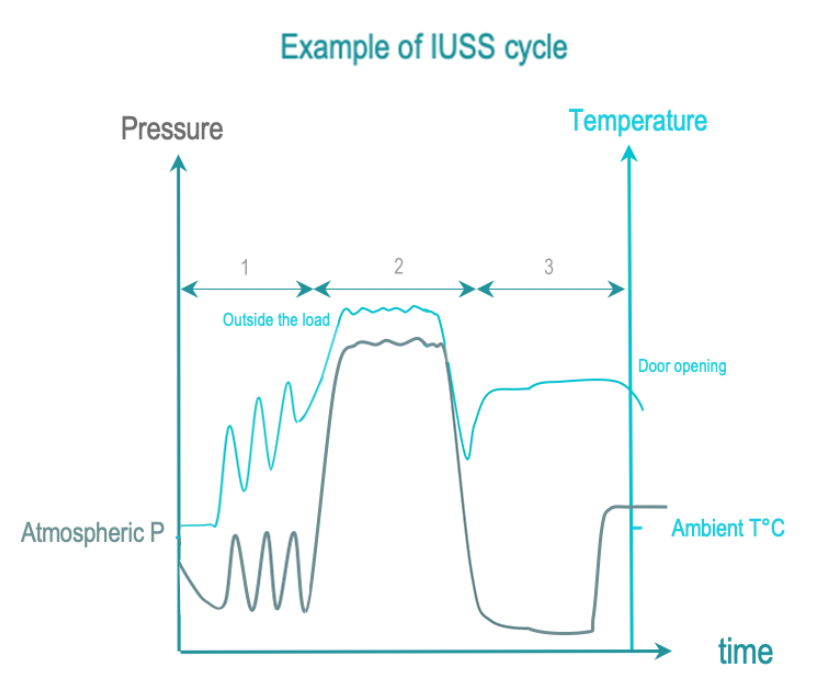
IUSS to be replaced by steam sterilization
Go to IUSS sterilization →
1 of 16 液体灭菌剂To be evaluated by WFHSS
Go to Liquid sterilization →
2 of 16 蒸汽Steam 134°C or 132°C preferred when allowed by RMD IFU
Go to Steam sterilization →
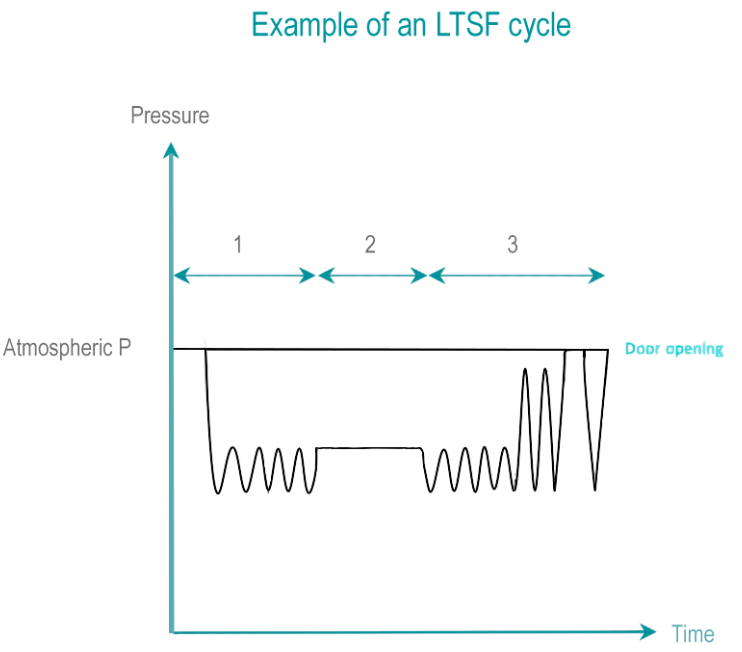
3 of 16 低温蒸汽甲醛Cycle according to RMD IFU
Go to Low temperature steam formaldehyde →
4 of 16 汽化过氧化氢Cycle according to RMD IFU
Go to Vaporized H2O2 →
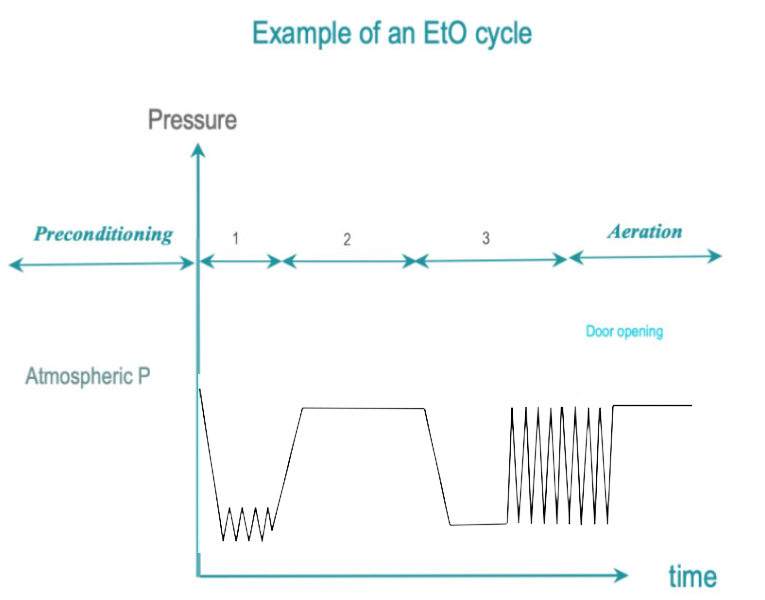
5 of 16 环氧乙烷Cycle according to RMD IFU
Go to Ethylene Oxide →
6 of 16 清洁、干燥、打包的 RMDNon packaged for non terminal sterilization
Go to choice of sterilization process →
7 of 16 无菌医疗器械Non packaged RMD for immediate use when non terminal sterilization
Packaged RMD for storage when terminal sterilization
Go to choice of sterilization process →
Terminal sterilization preferred to
non terminal
Go to choice of sterilization process →
9 of 16 非最终灭菌The RMD is not protected by a packaging and must be immediately used after sterilization
Go to choice of sterilization process →
10 of 16 +Terminal sterilization preferred
Go to recommendation of WFHSS for sterilization →
11 of 16 +Steam sterilization at 132°C or 134°C preferred when allowed by RMD IFU
Go to recommendation of WFHSS for sterilization →
12 of 16 +Visual control and routine controls
Go to Sterilization and quality management →
13 of 16 +According to RMD IFU
Go to choice of sterilization process →
14 of 16 +Steam sterilization at 132°C or 134°C preferred when allowed by RMD IFU
Go to recommendation of WFHSS for sterilization →
15 of 16 +Visual control and routine controls
Go to Sterilization and quality management →
16 of 16







