




The texts governing medical devices reprocessing and education activities may be grouped in 3 categories :
- Regulation (or legislation): Published by govermental bodies, they are of compulsory application. They can be national like in the USA1 or supra-national like in the European Union2. Example of key regulatory aspects pertaining to medical device reproccessing are the obligations for Medical Device Manufacturers to provide reprocessing instructions, for Healthcare facilities to report incidents related to Medical Device including those related to reprocessing. Regulation may also structure sterilization activities in a given country (for example, authorization to open a sterilization department, qualification required to Manage a CSSD, audit rules etc….), The present guidelines will not comment regulation.
- Standards (also referred to as norms): They set common expectations on products, services or processes to facilitate trade and enhance end user confidence and safety. Standards can be specific (for example steam sterilization processes or equipments) or transversal (for example, Quality Management). Standards are defined by consensus. Standards can be international (for example ISO), Regional (for example CEN for the European Union or national (DIN, BS etc..). National standards (i.e. applicable in only one country) will not be commented in this paragraph. ISO standards exist for sterilization but not for disinfection. Countries select the applicable standard for the various disinfection claim. (for example CEN and/or AOAC or ASTM)
- Standards are generally of voluntary application. Some can be made mandatory by local regulation. In practice, once a standard is adopted, it is convenient and safe for manufacturers, users and health care authorities to seek or verify compliance. When compliance is stipulated in a contract it is binding on the parties
- Guidelines (also referred to as recommended practices or best practices) are published by scientific societies, governmental bodies or standard organizations. Standards ease the interpretation of regulation and standards by healthcare facilities and serve as a framework for education programs and audits.
![]() At national level, guidelines are often co-written by 2 or more entities (for example Scientific societies and governmental bodies) or written by societies and endorsed by governments
At national level, guidelines are often co-written by 2 or more entities (for example Scientific societies and governmental bodies) or written by societies and endorsed by governments
In case of overlaps the applicable regulation always supersedes standards and guidelines.
![]() In the case of the European community, the compliance to an harmonized standard (i.e. a standard endorsed by the CEE) indicates that the essential requirement of the medical device regulation are met. Standards are also used as a reference by notified body for CE marking.
In the case of the European community, the compliance to an harmonized standard (i.e. a standard endorsed by the CEE) indicates that the essential requirement of the medical device regulation are met. Standards are also used as a reference by notified body for CE marking.
![]() Standards are subject to copyright and fees. Revenues generated by the sales of standards are used to finance the standardization effort and organizations.
Standards are subject to copyright and fees. Revenues generated by the sales of standards are used to finance the standardization effort and organizations.
There are many standards but not all products or processes are covered by standards. For example there are ISO and EN standards for automated cleaning and disinfection processes but none for manual processes.
The figure below illustrates ISO and CE framework for sterilization activities. It is provided for illustration purpose only. This diagram is subject to constant change and only valid for the indicated period. Official list and status can be found on ISO and CEN websites
![]() Radiation sterilization standards which are not commonly used in routine in healthcare facilities are not represented.
Radiation sterilization standards which are not commonly used in routine in healthcare facilities are not represented.
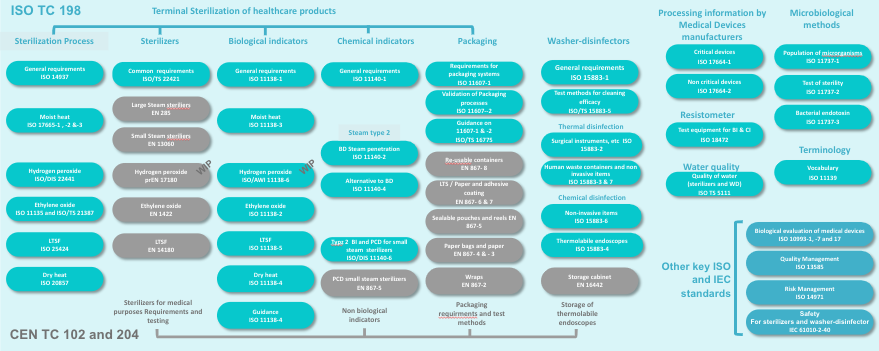 | 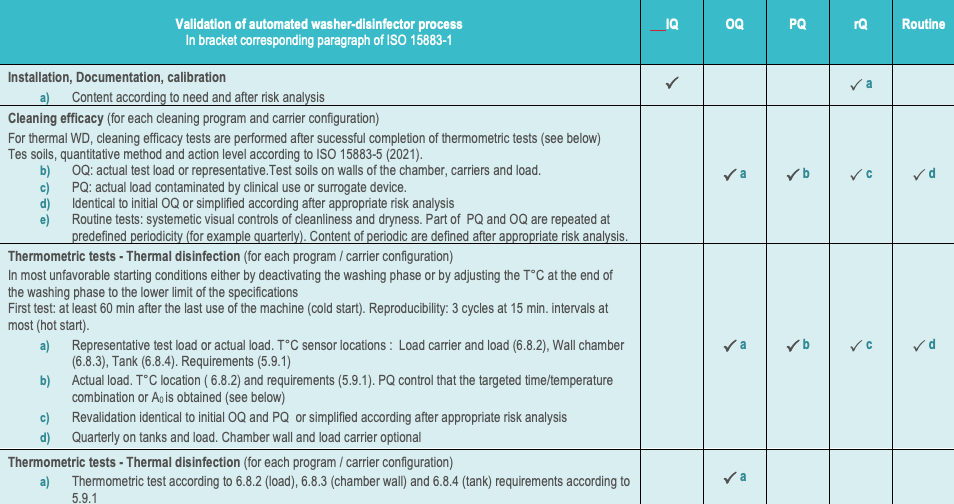 |
ISO standards for thermal and chemical washer-disinfectors 35,36,37,38,39 are also primarily intended for manufacturers. However they contain guidance for healthcare facilities to check the conformity of installed washer-disinfector throughout its working life. ISO 15883-440 and EN 1644241 are dedicated to heat sensitive flexible endoscopes (reprocessing and storage respectively). ISO 15883-542 describes test method and criteria for cleaning efficacy
![]() EN sterilization standards are often referred to as equipment standards and ISO standards as process standards. This is not an official ISO language but it well depicts the respective role of each group of standards.
EN sterilization standards are often referred to as equipment standards and ISO standards as process standards. This is not an official ISO language but it well depicts the respective role of each group of standards.
Other ISO TC 198 standards and guidance displayed on the above diagram come in support of other TC 198 documents.
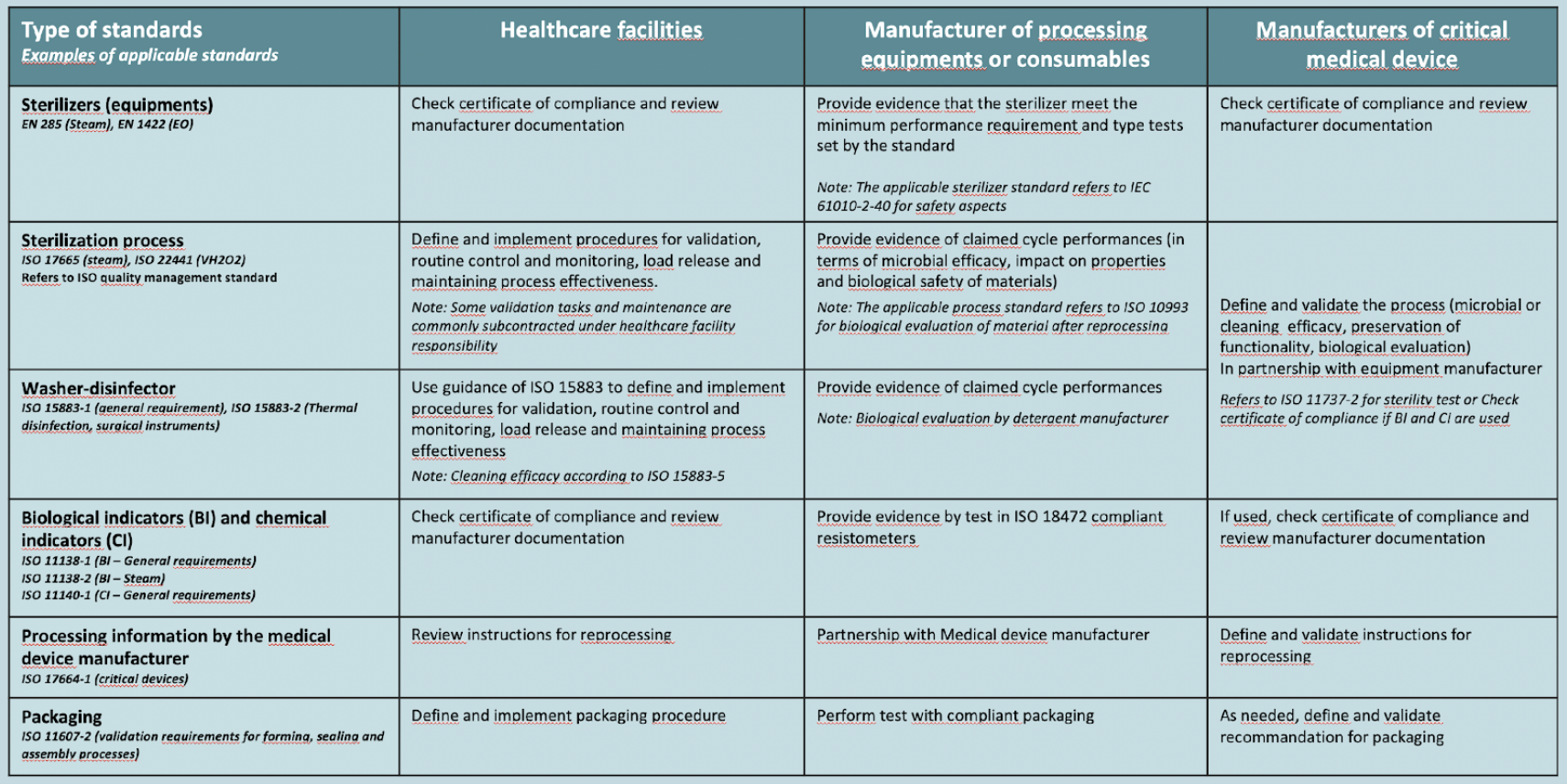
![]() An example of a difference between healthcare facilities and device manufacturing industry are loads configurations: Typical single use medical device manufacturer loads are homogenous (i.e. same single use item with initial contamination levels under control. Typical healthcare facility load are heterogenous (i.e. different devices with variable levels of contamination even after thorough cleaning)
An example of a difference between healthcare facilities and device manufacturing industry are loads configurations: Typical single use medical device manufacturer loads are homogenous (i.e. same single use item with initial contamination levels under control. Typical healthcare facility load are heterogenous (i.e. different devices with variable levels of contamination even after thorough cleaning)
- Standard must be compatible with local guidelines or regulation. For example, responsibility assignments (who does what) are considered as guidelines or regulation matters are not addressed by standards.
![]() Some standards provide informative (non normative) guidance that will describe responsability assignements for various scenario such as healthcare facility.
Some standards provide informative (non normative) guidance that will describe responsability assignements for various scenario such as healthcare facility.
- Among the standards of highest level of interest to user there are the sterilization process standards. The table content of those standard is aligned on ISO 14937.
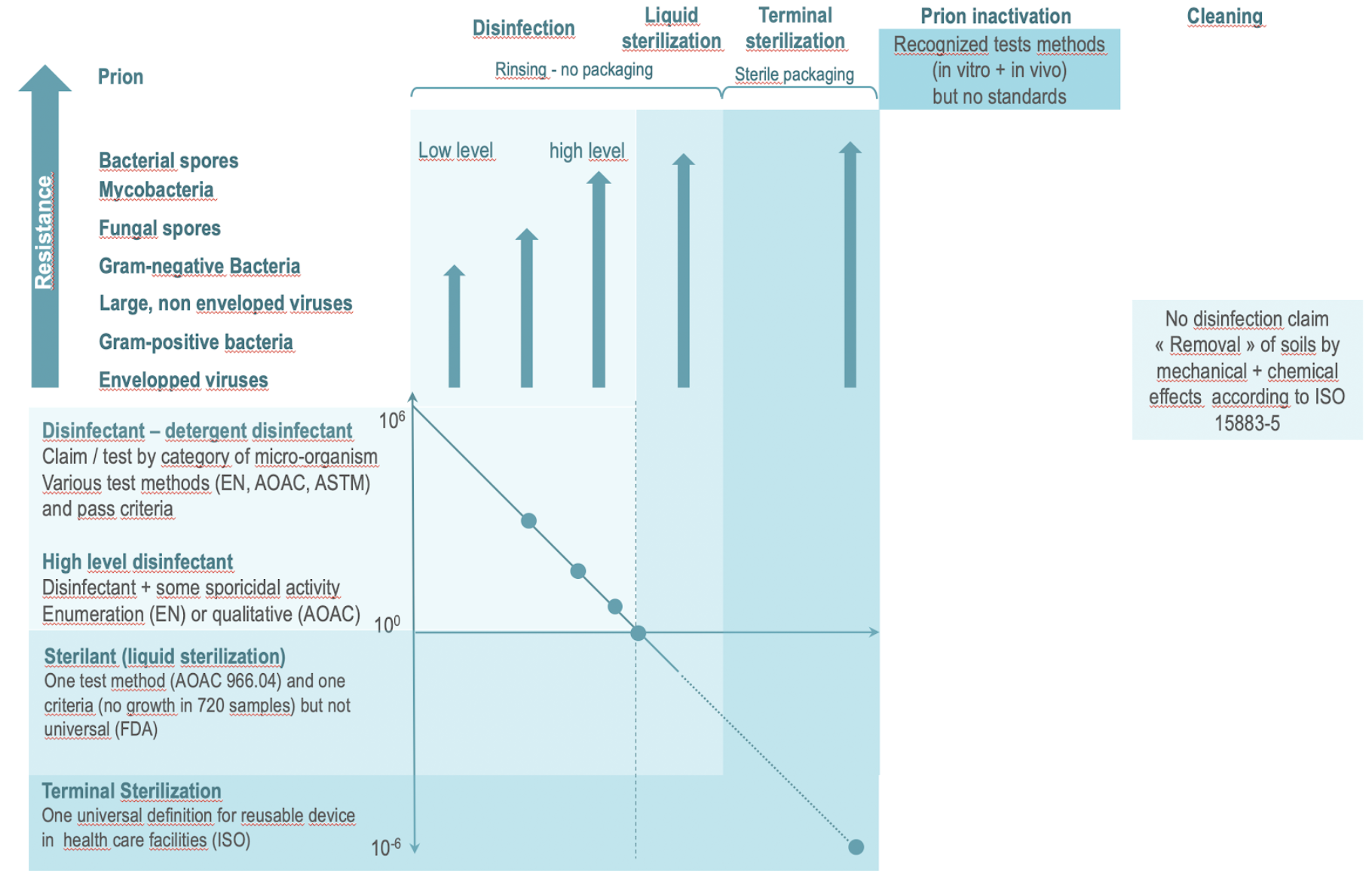
Unlike for sterilization, there are no universal ISO Standards for evaluation of disinfection chemicals. Depending on the region different methods and acceptance criteria apply.
![]() For example EN standards in Europe, AOAC or ASTM in USA. Some countries may look at both EN and AOAC and ASTM, in some cases, with adaptations (for example Australia)
For example EN standards in Europe, AOAC or ASTM in USA. Some countries may look at both EN and AOAC and ASTM, in some cases, with adaptations (for example Australia)
Disinfectant are commonly classified in 3 categories: low level, intermediate level and high level. However the borders between each category are not universally defined. For example high level disinfection is always associated to some level of efficacy on spores but the efficacy threshold on spores is not the same.
Unlike for sterilization in health care facilities, where sterility test are performed only with the germs predetermined as the most resistant, disinfection claims are for specific categories of germs : bacteria, fungi and yeast, virus (envelopped and non envelopped) and mycobacteria and spores, expressed in log reduction, with log reduction objective which may differ between catgories of microorganismes and region.
![]() For sterilization in healthcare facilities according to the overkill principle: > 6 log of the spore selected as the most resistant to the sterilization process and SAL of 10-6
For sterilization in healthcare facilities according to the overkill principle: > 6 log of the spore selected as the most resistant to the sterilization process and SAL of 10-6
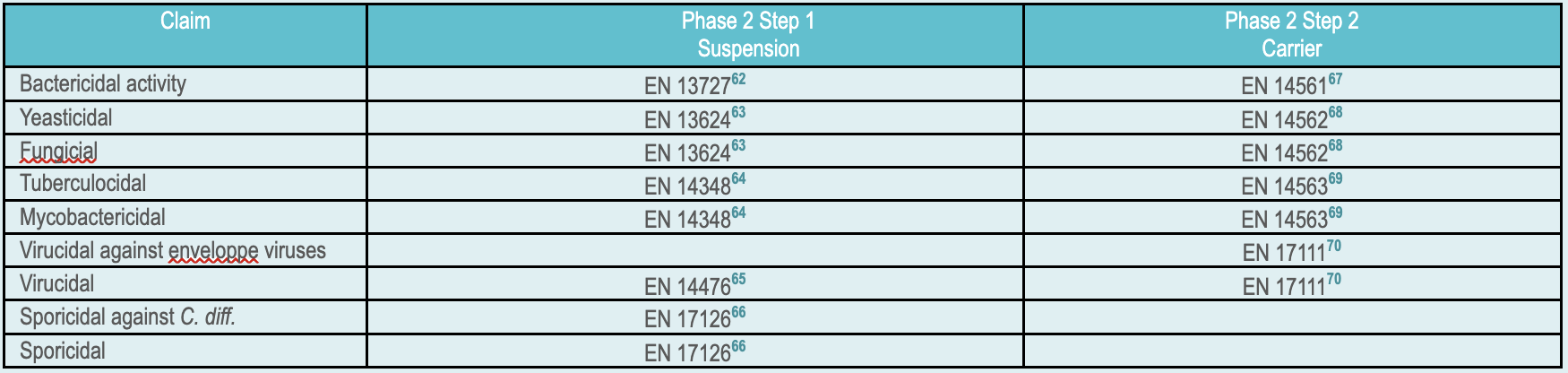
In the European community the test method to be applied for the various type of disinfection claims are defined by EN 14885.
EN 1488561 covers human medicine, veterinary, food, industrial, domestic and institutional areas. Within the medical domain EN 14885 proposes adapted substantiating strategies for various field of application hand hygien (hygienic handrub, hygienic handwash, Surgical Handrub or-wash), surface disinfection (with or with mechanical action and airbone), textile disinfection and aqueous system and instrument disinfection. The rest of this paragraph will focus on disinfection of instruments. EN 14885 list test in suspension (phase 2.1) or on carrier (Phase 2.2). For medical devices, phase 2.2 carrier tests are more representative of real life and closer to AOAC and ASTM approach. Interference matter is added to simulates soil (in clean or dirty condition depending on the application) Test are performed at the concentration and exposure time specified by the manufacturer. At the end of the exposure time a neutralizer is added to stop the microbiocidal activity before counting to check that the targeted log reduction is achieved.
|
Unlike for disinfectant there are no international standard for the evaluation of cleaning efficacy but the efficacy of the cleaning process (i.e. combination of mechanical and chemical action) can be evaluated according to ISO 15883-5.
![]() Although ISO 15883-5 was developped for the evaluation of automated process. It may be applied to manual processes with the drawback, for manual processes, that there are operator dependence.
Although ISO 15883-5 was developped for the evaluation of automated process. It may be applied to manual processes with the drawback, for manual processes, that there are operator dependence.
 |








